大家上午好,我是苏晓泉,来自青岛大学计算机学院。非常高兴今天能够跟大家分享一下,我们开发的基于搜索的微生物组研究的新模式。
首先介绍一下我们的团队——青岛大学生物信息研究组,致力于高性能的生物信息方法学研究。
尤其是在微生物组相关领域,我们已经开发了和维护着一系列的自主知识产权的软件和工具,包括微生物组分析套件Parallel-Meta Suite、微生物组距离的系列算法Meta-Storms、微生物组搜索引擎MSE(Microbiome Search Engine),以及微生物组的功能校正算法Meta-Apo等等。
跨学科困惑
生物信息,已经融入到了研究和应用中的各个方面。然而,相关工具和软件的开发,其实并不容易!
我是纯计算机科学背景的,硕士毕业之后才开始从事生物信息和微生物相关的工作。所以在跨学科的过程中,除了思路上要转变之外,还要学习很多的新知识和新的概念。
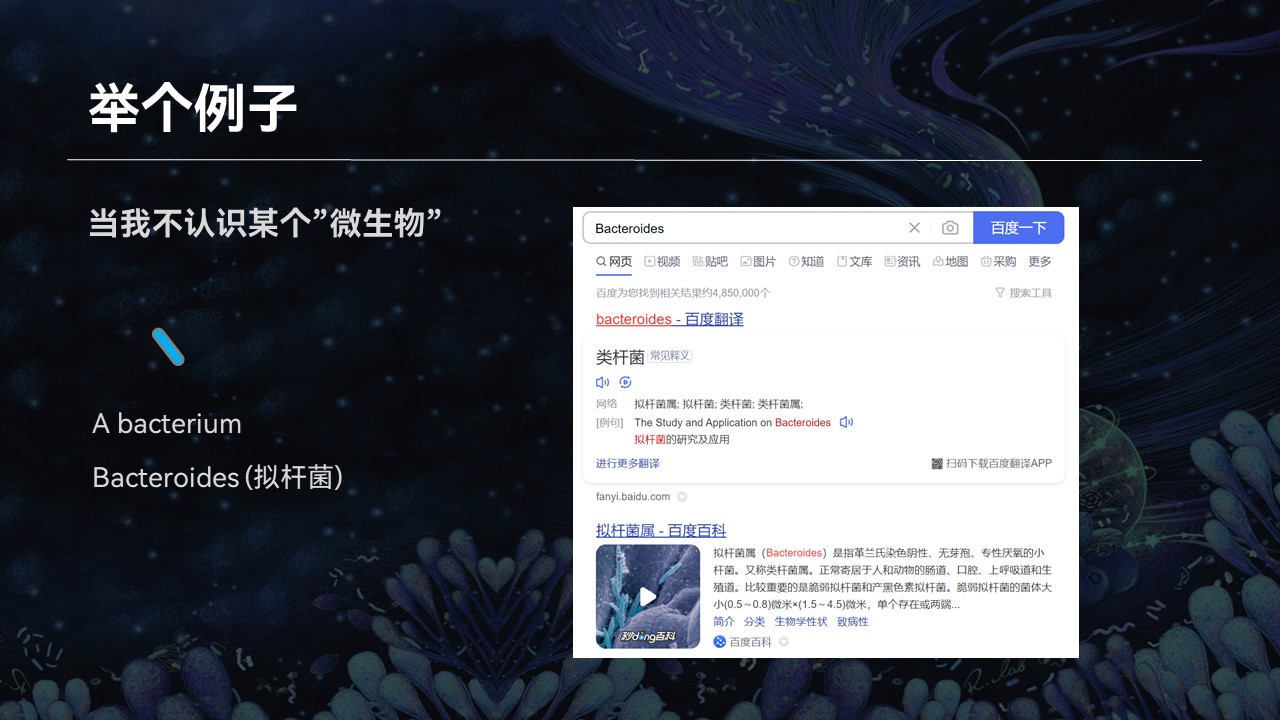
但是当我在阅读相关资料和文献的时候,有大量的知识点和词汇,我不认识、不知道,给我带来了很多的困扰。
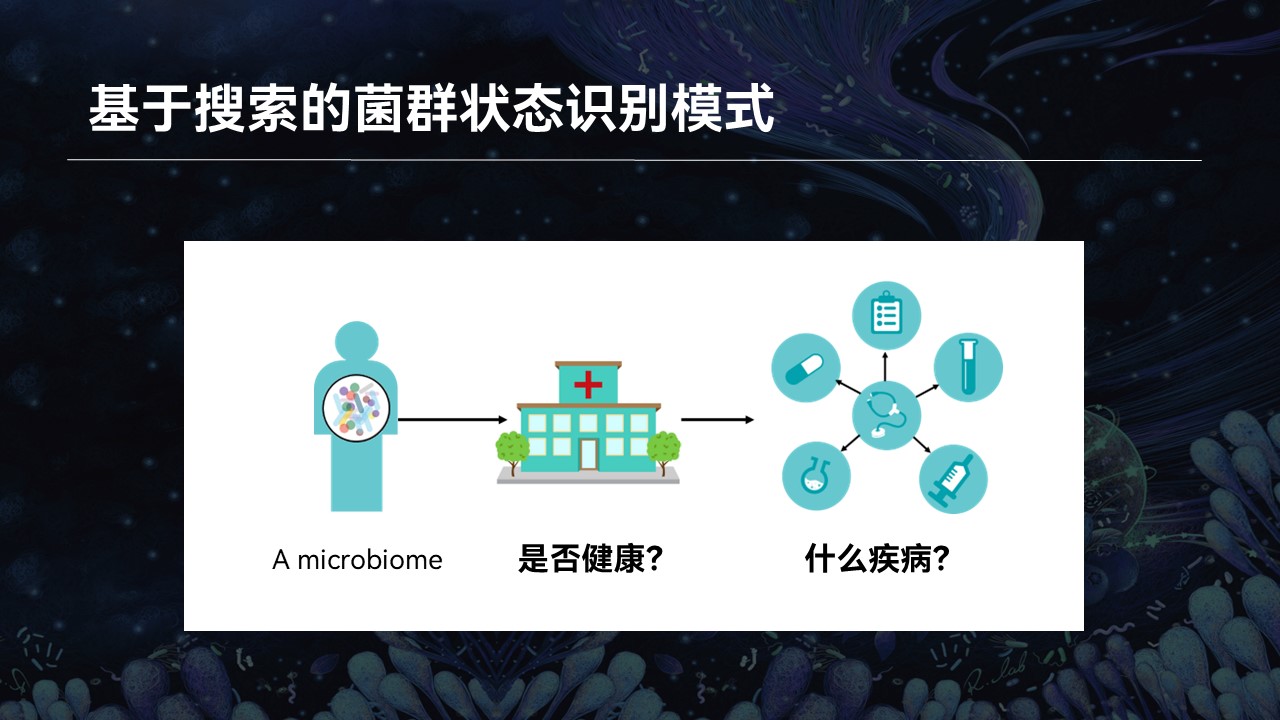
比如说一个生物学名词,我不认识,那么我会去网上去查找它相关的资料。比如在搜索引擎中输入这个名词,网页的搜索引擎就会根据关键字的匹配返回相应的搜索结果,我也就会知道这个微生物它是什么、有什么作用、什么来源等等。
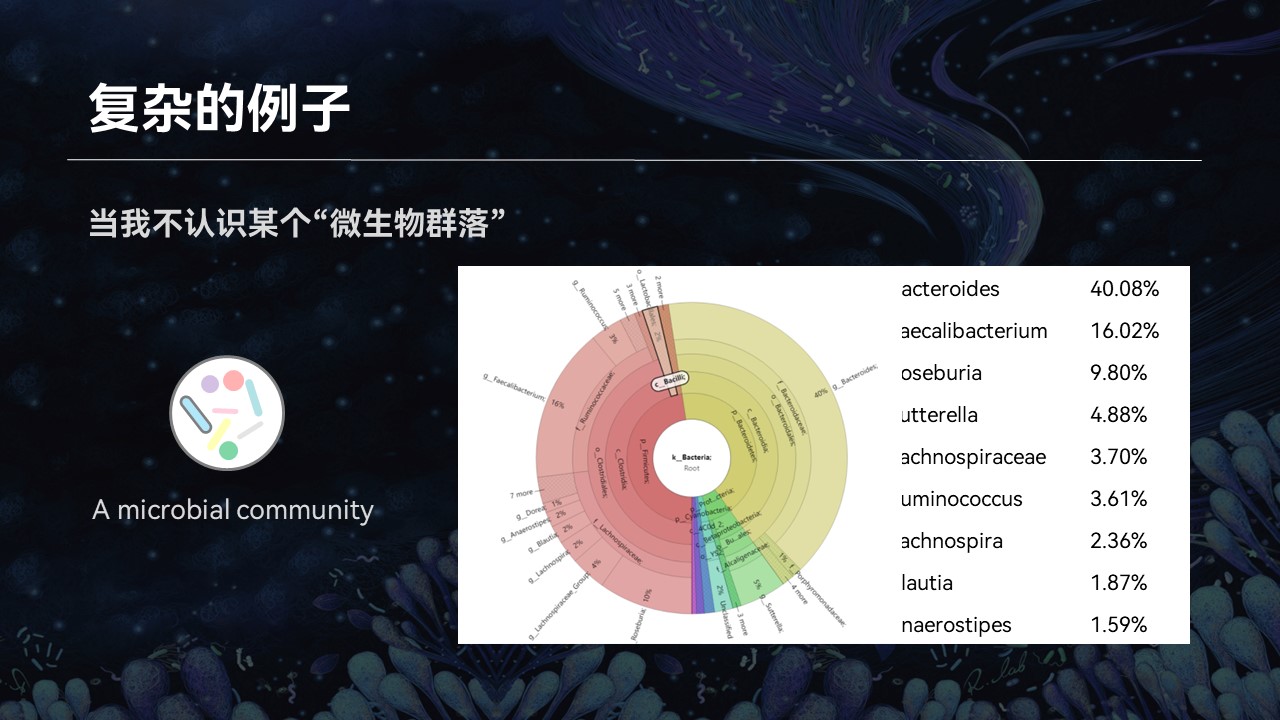
然而后来我接触到了微生物群落,我意识到了事情其实并没有那么简单。
因为一个复杂群落中可能还含成百上千种不同的物种,我几乎不可能将每一个名词都在网上搜索一番。即便是我认识里边每一个物种,它们组合在一起是什么意思、这个群落有什么特征、有什么特性、来源如何……我也不得而知。
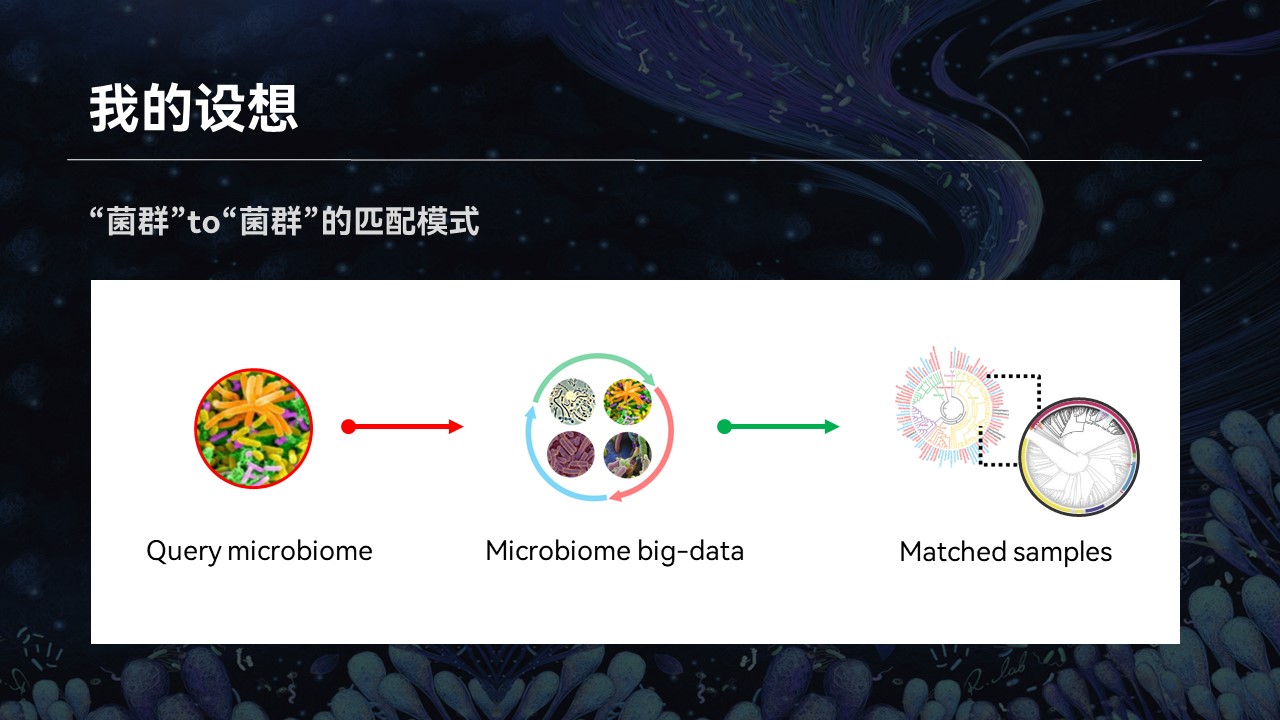
那么这个时候,我又有一个设想:如果我们能够利用当前已有的群落的大数据,根据群落的整体结构进行搜索和匹配,来做到一个菌群对菌群的匹配结果,返回的信息将会帮助我们认识和了解微生物群落,也可以推动相关的研究与进步。
见招拆招
有了目标有了想法之后,我们面临着一系列的实际的问题。
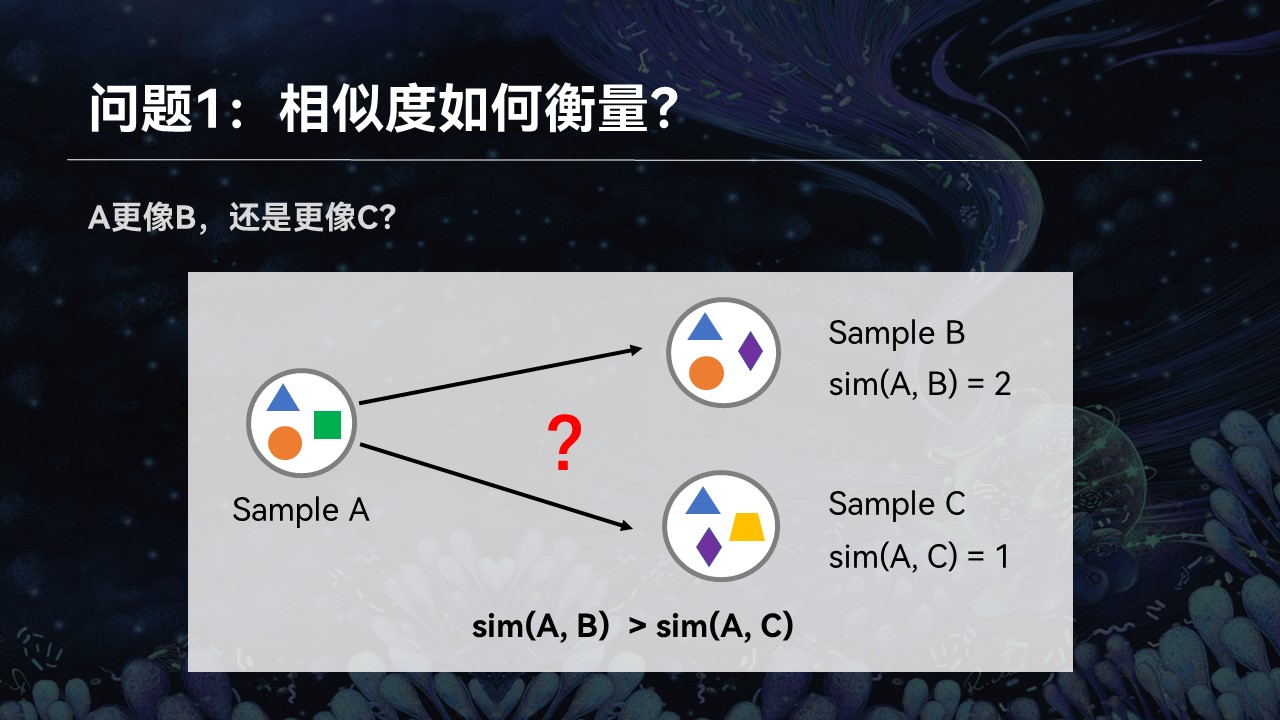
Q1:相似度如何衡量?
第一个就是对于复杂的群落来讲,我们如何来量化地评价它们之间的匹配性和相似性。
通常,我们目前研究菌群的最主要的途径就是测序,比如扩增子、宏基因组、宏转录组等等。对于短序列的分析之后,我们可以获得群落的特征,像分类单元、物种名称、功能基因等等。因此,我们可以利用这些特征来去量化衡量群落之间的匹配程度。
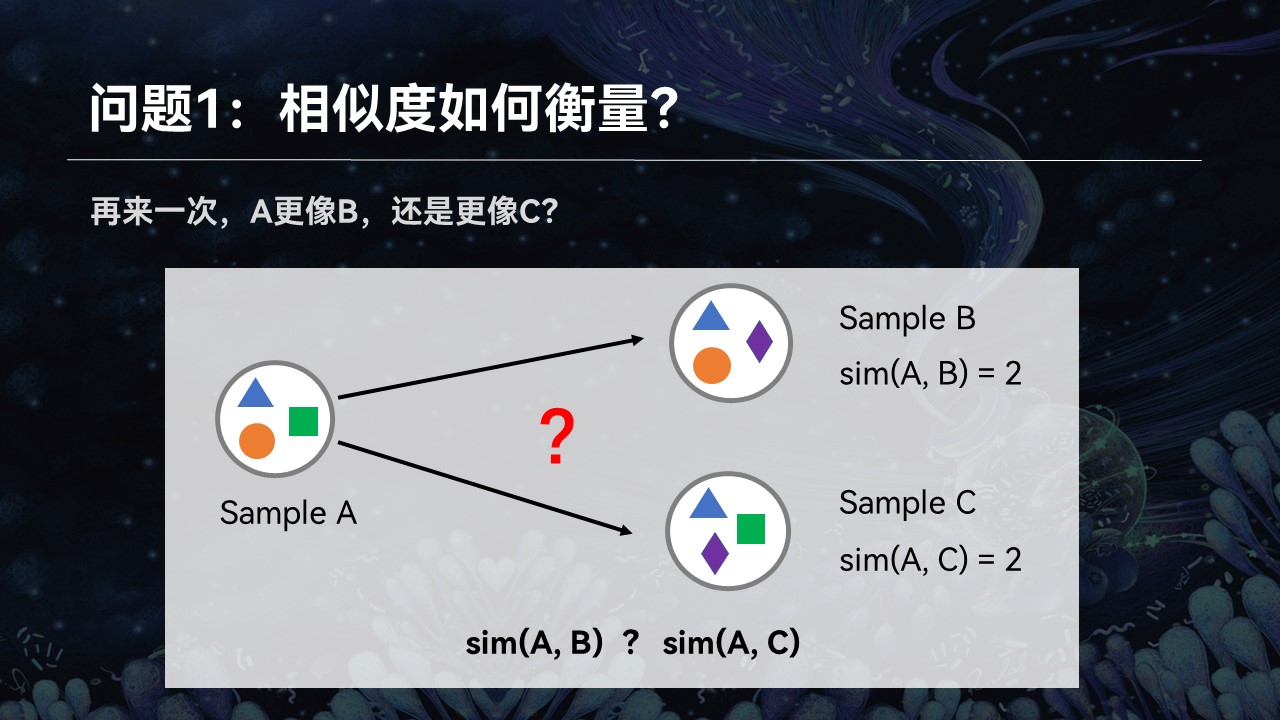
举个例子来讲,我们这儿有3个群落A、B、C。我们想知道样品A更像B还是更像C,它跟谁的匹配程度更高一点。很简单,通过观察我们可以看到,A、B之间共享两个物种,而A、C之间只有一个物种是共同的。我们可以简单地认为,A、B之间的匹配程度更高一点。
让我们把这个例子稍微做一些修改,让A、B与A、C之间的共享的物种数量差不多,刚才的方法就不适用了。我们也意识到,单纯通过统计学、数学和算法,不可能解决生命科学中面临的问题。
所以这个时候,我们就额外引入了物种之间的亲缘关系,来进一步地去从精细层面量化它们之间的差异。
进一步的观察我们发现,即便是AB与AC共享的物种数量非常接近,但是我们可以看到,AB之间存在差异的物种,在亲缘关系上处于相同的分支,而AC之间的差异更远一点。所以我们仍然可以得出结论:AB之间的匹配程度更高一些。
那么利用以上的想法,我们在2012年就开发出了基于进化的微生物组之间的相似性算法,并且后续陆续也开发了面向分类单元、物种以及功能结构等等不同特征的算法。
近期,我们也刚刚研发了只利用部分特征来进行局部匹配的算法,可以通过不同的特征、不同的角度,来全面衡量复杂群落之间的相似性和匹配性。
有了度量算法之后,我们本以为可以实现刚才的想法,但是摆在我们面前有一些新的问题。
Q2:如何快速匹配?
因为随着微生物组的广泛推广和测序技术的进步,以及成本的降低,海量的数据被生成了。
据不完全统计,在NCBI、EBI以及Qiita等公开数据集中,已经存在了超过百万量级的微生物群落样品。那么在如此浩瀚的数据海洋中一个一个地进行匹配,要同时考虑到物种的相似性、物种的组成丰度,以及它们之间的亲缘关系,犹如大海捞针一般的困难。
这个计算的量和复杂程度是我们无法承受的。
所以我们继续去研究开发了,面向于复杂群落的动态索引的策略。
举个简单的例子,我们有一本非常厚的字典,想在这个字典中找到我们想要的那个字,如果我们一页一页地去翻,很困难找到。但是当我们利用这个字的偏旁部首、利用它的读音等特征进行索引,那就可以很快地找到。
同样的道理。对于海量的复杂群落,我们首先进行降维处理,将它复杂的特征简化和压缩为一些代表性的索引特征。当有新的样品进来的时候,我们先利用这些索引特征进行简单的查找,选取一些疑似性的候选匹配者。
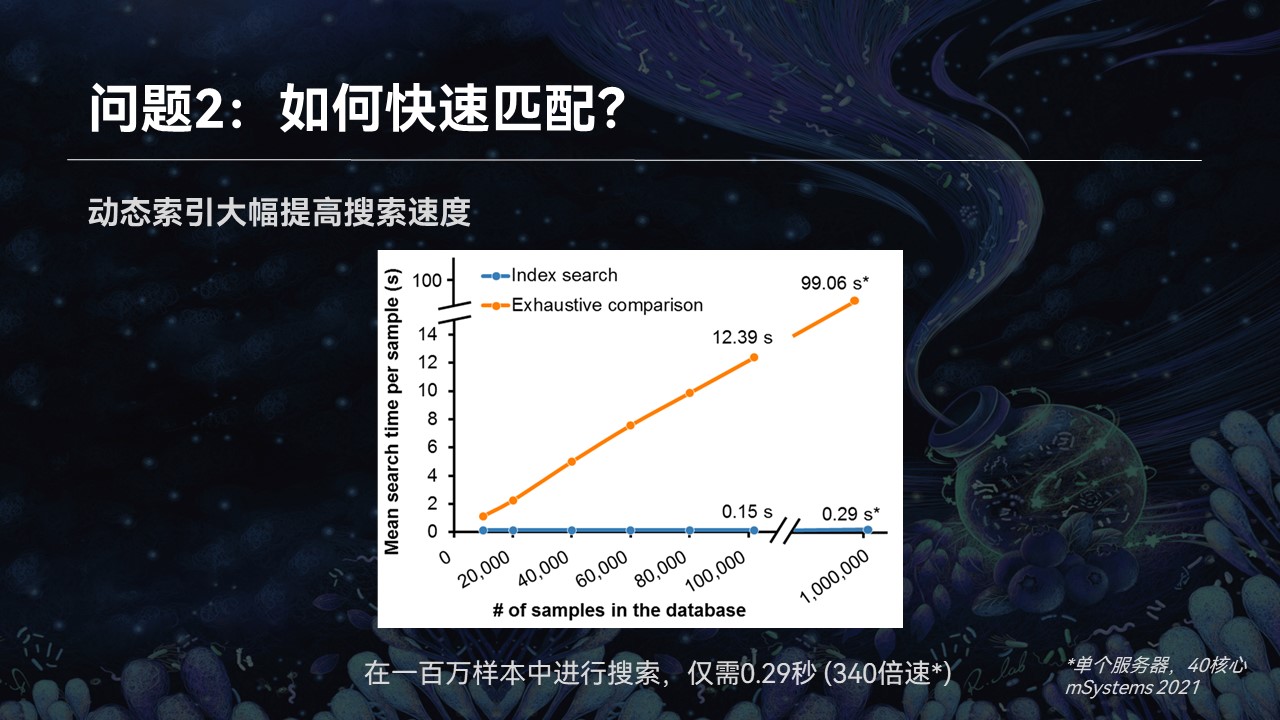
由于索引的特征维度较低,很简单,所以可以在很短的时间内就可以大幅降低我们的搜索范围。接下来我们就可以在为数不多的候选者中,利用群落完整的特征,进行精确的查找匹配,最终返回我们的搜索匹配结果。
同时在这个过程中,我们也加入了多线程的并行计算、内存共享等等计算优化技术,使得在100万样品的搜索过程中,在单个服务器上不到0.3秒就可以完成仓储的过程。
此外,由于我们索引的稳定性,即便是数据量膨胀10倍,搜索时间也不会发生明显的改变。这就为我们在海量的数据中进行数据挖掘提供了基础。
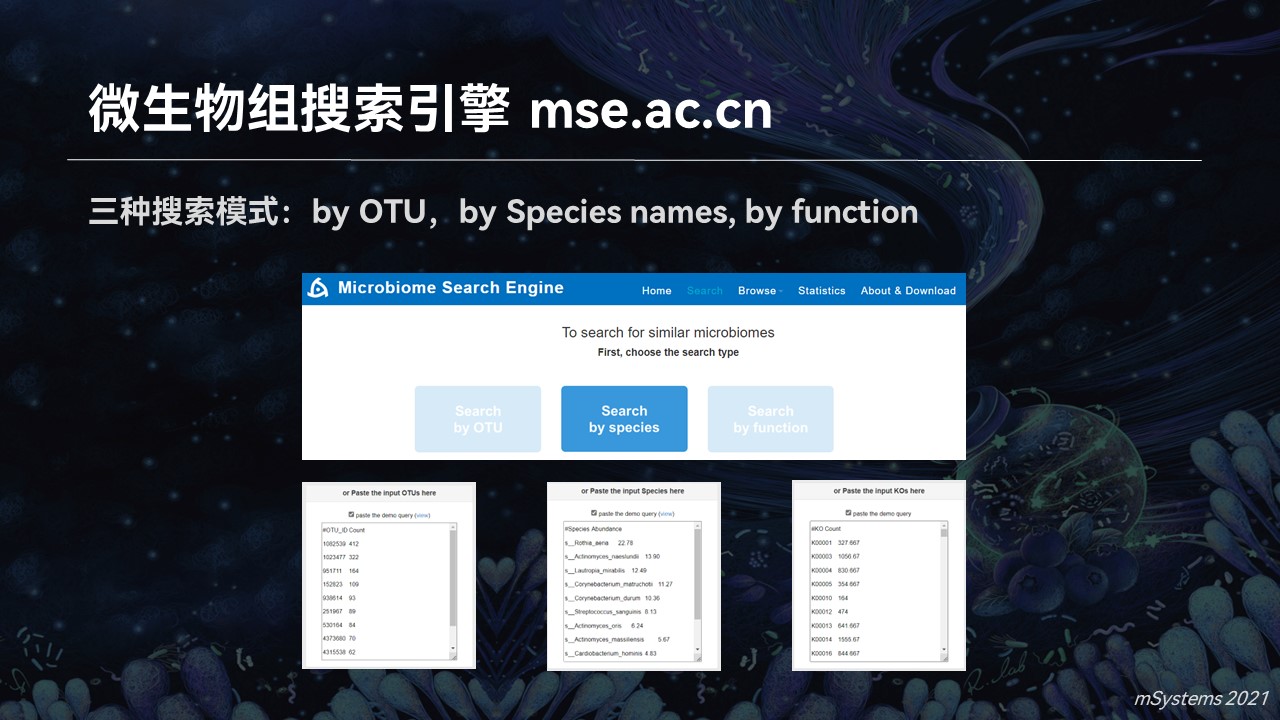
因此我们前段时间上线了首个微生物组的搜索引擎,也让我之前的畅想成为了现实。
目前的搜索引擎中,支持分类单元、物种名称以及功能基因等不同的搜索模式,可以完成群落对群落的匹配结果。
这样的匹配结果中,也包含着匹配到的样品,它们的来源、量化的相似度以及可视化的匹配的结果展现等等丰富的信息。
这些信息的原始数据也都可以下载,供以后更加深入的分析和挖掘,比如说物种的来源追踪、菌群状态的识别等等。
除了算法之外,我们的搜索引擎也离不开数据的支持。
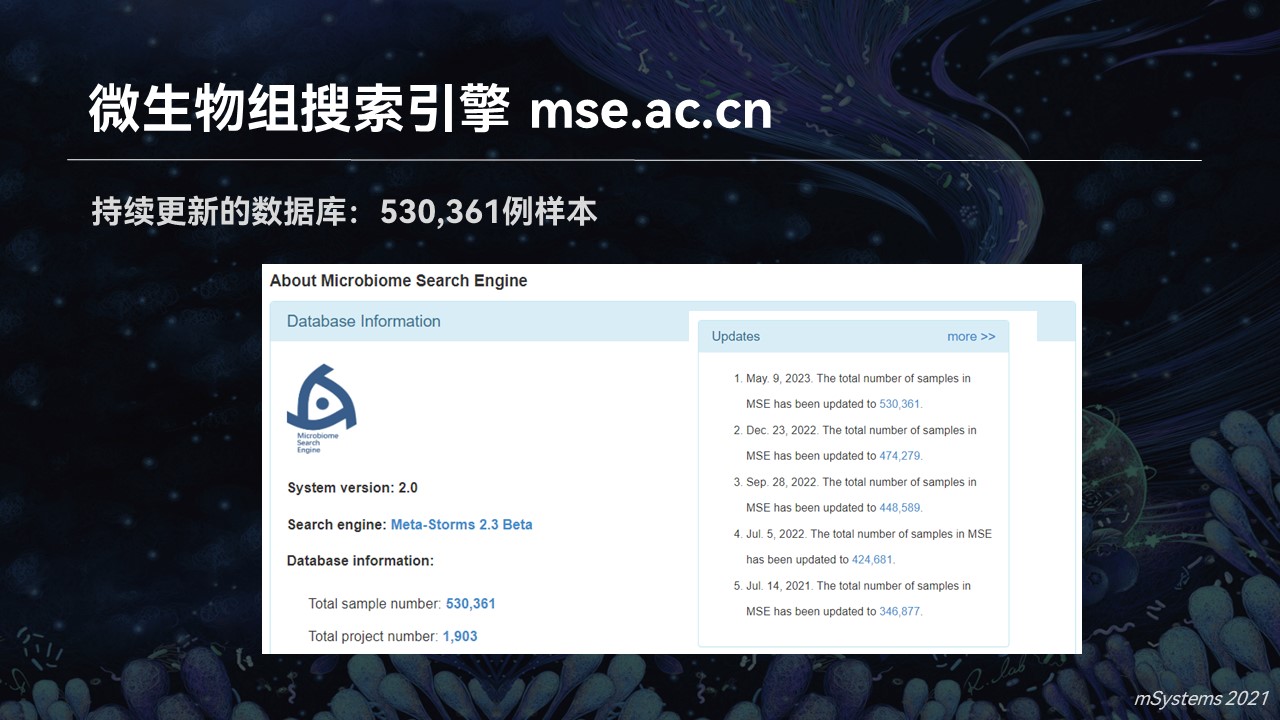
从最初的几千例样品,经过多年的不停的累积,目前搜索引擎中已经包含了超过53万个高质量的微生物群落样品。它来源于1900多个不同的研究和课题,其中包含人体肠道、口腔、皮肤、生殖道以及海洋、土壤、动植物等等多个生态系统和来源,是目前质量最高、数据量最大以及涵盖了生态系统最完整的微生物组数据库之一。
我们所有的样品都经过了统一化的处理、人工的质量控制和筛选以及标注等等。
新的发现
有了海量的数据,有了先进的算法,我们似乎可以进行一些脑洞大开的尝试。
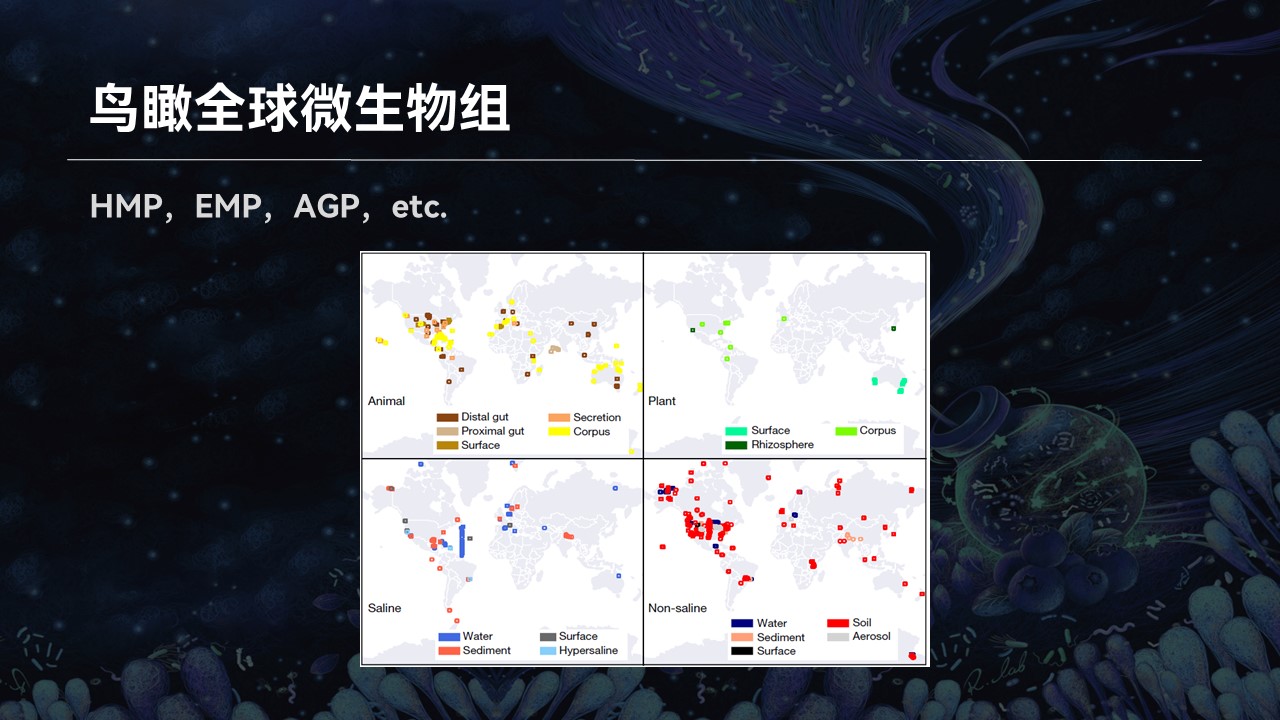
在过去的几年中,地球微生物组计划、人类微生物组计划以及美国肠道计划等等产生了海量的样品,为我们的研究打开了一扇新的大门。
如此大队列的研究,不禁引发了我们新的思考,就是在地球各种各样的生态系统中微生物群落的组成分布有什么规律?
因为从简单的角度来考虑,一个复杂群落都是由不同的微生物物种混合在一起共生。如果我们从进化树中随机抽取不同的物种混合在一起,产生一个新的组合,那么从理论上来讲,这样的组合种类有无穷多的可能性。
然而事实上真的如此吗?是不是有无穷多种不同类型的微生物组存在?
为了回答这个问题,我们根据搜索引擎的匹配结果设计了一个微生物组的“新奇指数”,从而判断一个新样品的独特性。
很简单,当有一个新的样品产生的时候,我们去已有的数据库中进行搜索匹配。如果它匹配到的菌群跟它的结构都非常相似,这说明类似的群落结构我们以前已经见过了、采样过了,在我们的数据库中已经诞生过了,它的菌群结构新颖度就不是很高,它的新奇指数(MNS)就比较低。
反之,即便是一个新菌群,它的最佳匹配结果跟它的相似度都很低,说明类似的菌群结构并不常见,它的新奇指数就非常高。
因此,我们把搜索引擎中所有样品连续多年的新奇指数进行了一个追踪,发现了一个非常有意思的现象:在过去很多年中,我们的样品总数不断地在攀升,然而其中人体菌群的高新奇指数的样品数量却没有发生明显的变化。
换句话来说,我们一些新的样品,它的群落结构其实之前我们已经见过,而且大部分样品都是这样子的。
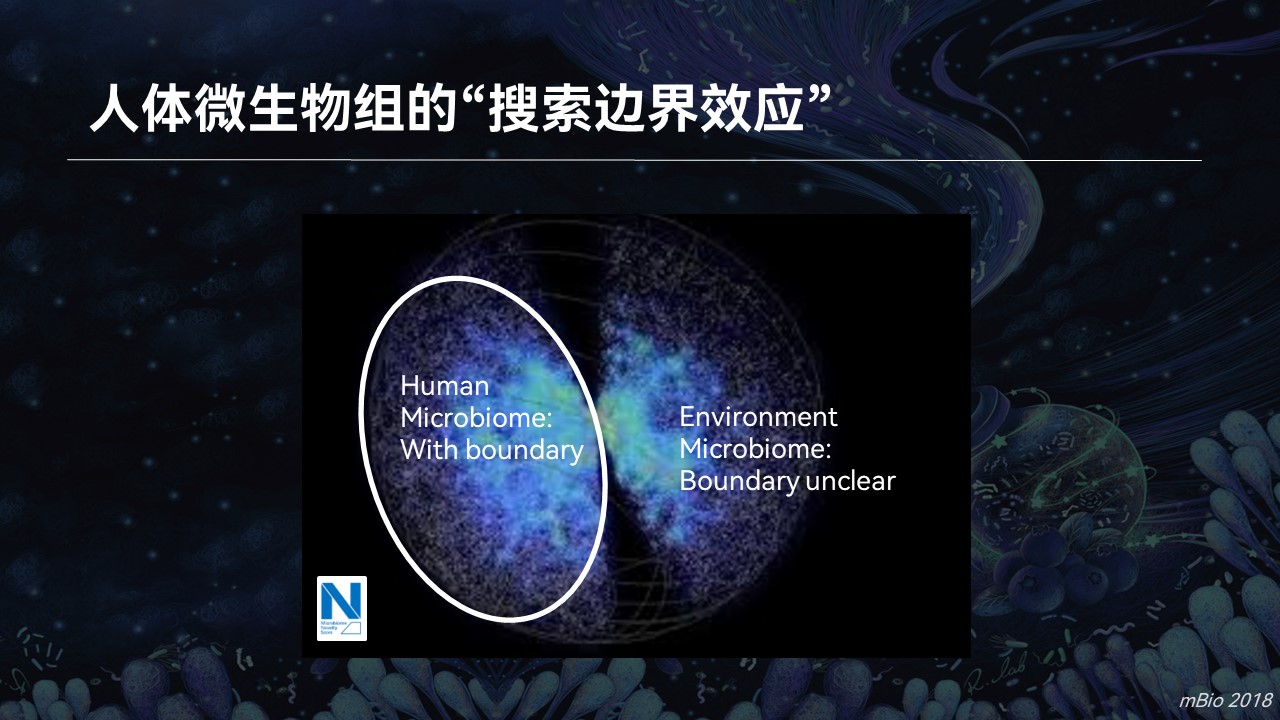
如果我们放眼到整个微生物组的数据空间当中去,利用搜索引擎,我们会发现即便是人体菌群,个体之间差异很大,多样性也非常高。在搜索模式下,它们总是处于一个边界当中在不停的变化。我们把这种现象称之为人体微生物组的“搜索边界效应”。
而对于环境菌群,这种边界并不明显。
新的应用
这样的一个抽象的发现对于我们实际的研究和应用,又有什么帮助呢?
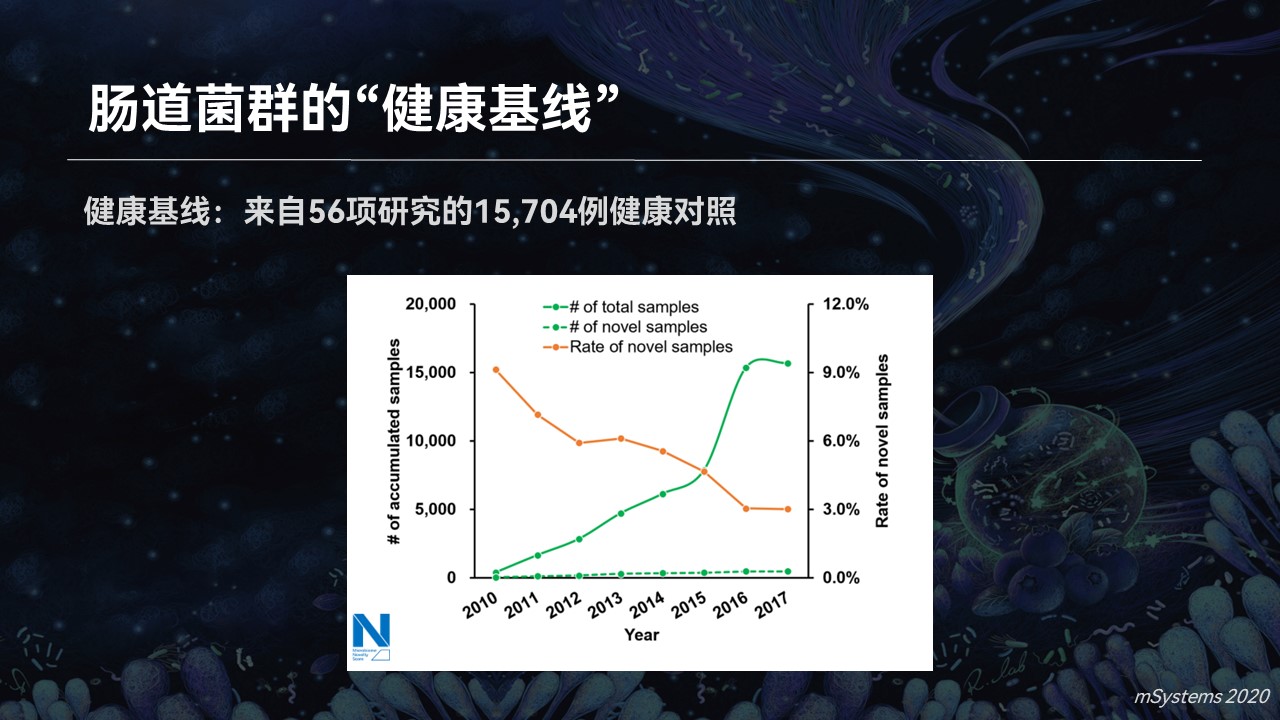
当我们把眼光再次聚焦到肠道菌群中去,我们仍然发现了肠道菌群的一种搜索边界效应。也就是说,对于健康的肠道菌群,它的总数一直在升高,然而其中新奇的样品数量并不会一直增多,会趋于一个稳定的状态。
这就好比地球上有各种各样的不同的人,我们的长相、身高、体重都不一样。但是当我们见多识广,见过了形形色色的人之后,我们突然遇到一个外星人,那见多识广的我们可以一眼都判断出来,外星人不属于我们地球,不是我们地球的人种。
同样的道理。当我们累积了足够多的健康肠道菌群之后,我们可以建设一个菌群的健康基线。那么在筛查当中,我们是不是就可以一眼就能判断出哪些不属于健康基线,它是状态异常的样品。
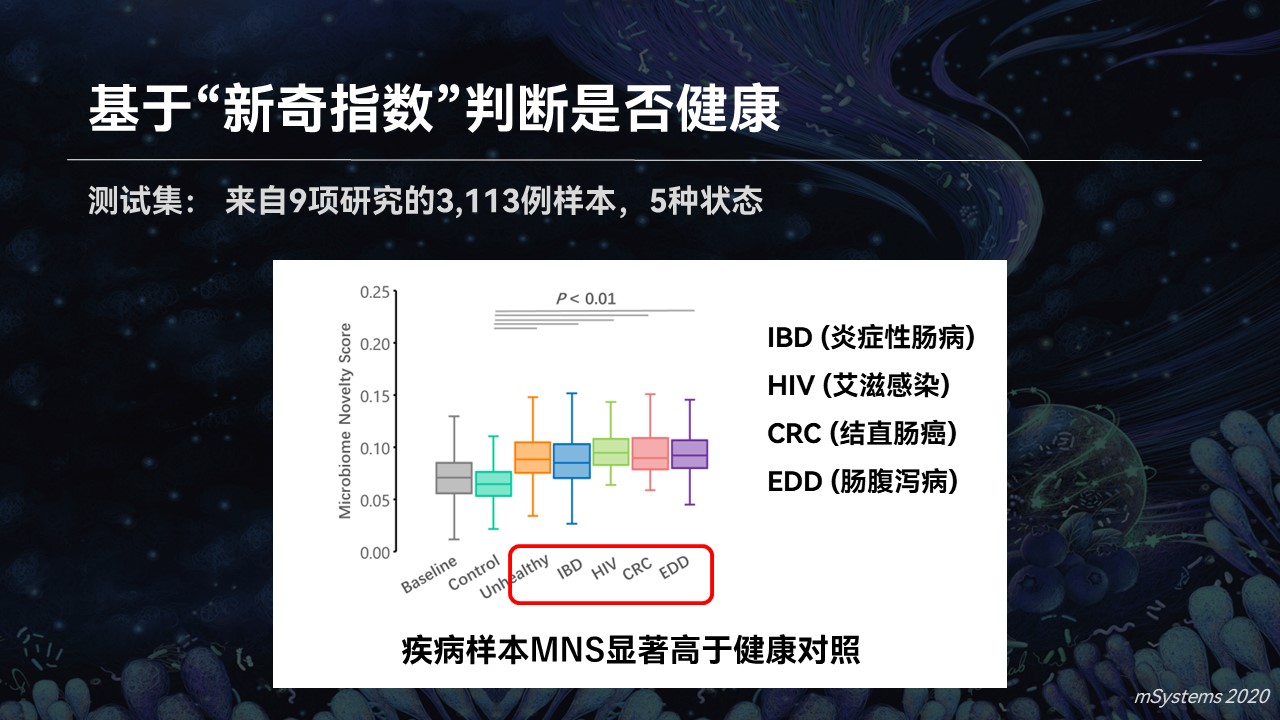
我们也用一个试验进行了验证,相对于一个超过1.5万例健康基线的数据库,疾病样品的新奇指数要显著高于它们的健康对照。
而且在多种疾病中都观察到这个结果。所以利用搜索引擎和新奇指数,我们可以成功地去判断出哪些样品的健康状态是异常的。
但是这种方法、这种结果对于我们常用的机器学习分类器来说,是很难做到的,因为它必须依赖于健康对照和具体的疾病样品来建立模型。
之后,我们就要对那些状态不正常的异常样品的具体疾病类型进行分类识别。
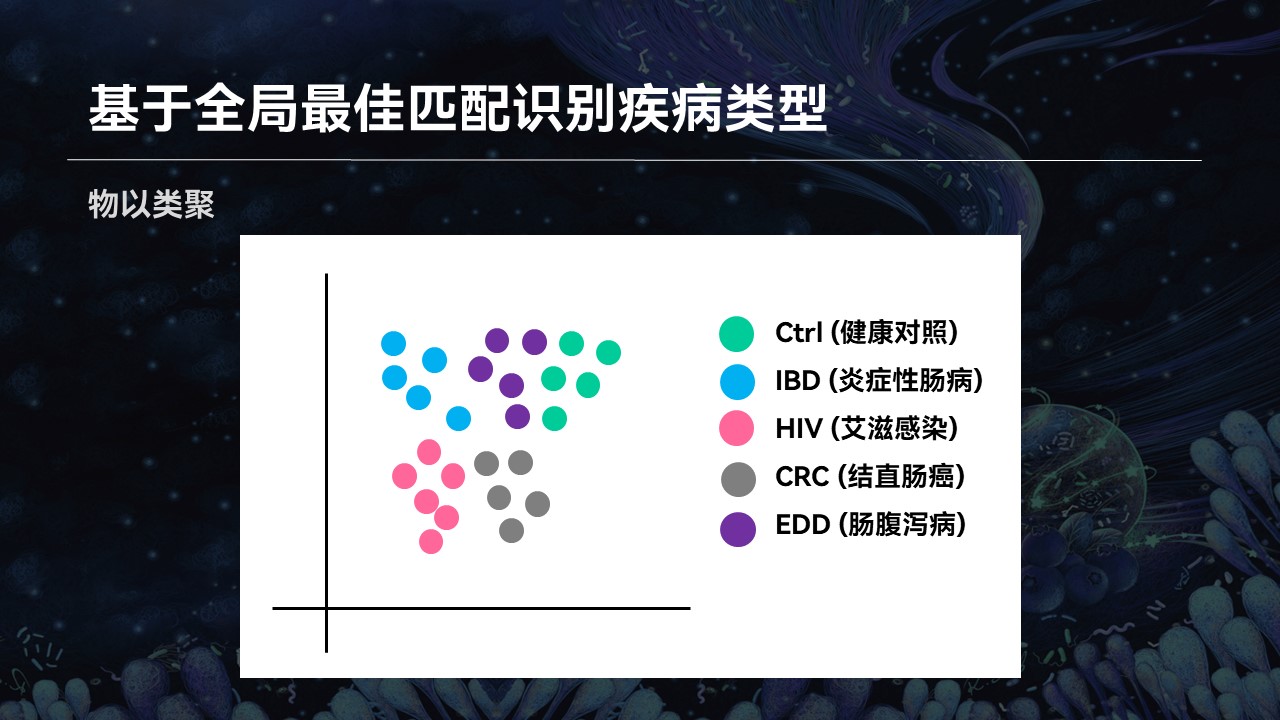
大家可能在很多文献中,见过有类似的这种聚类的图。不同的疾病类型,菌群可以被聚在一起,它们会物以类聚。
所以我们也利用这个现象,使用搜索引擎进行匹配。如果说一个样品的匹配结果大部分都处于某种状态下,属于某种疾病类型,那么这个样品也可能属于同样的类型。
我们发现,基于搜索的方法在具体疾病类型判断上,整体的效果也是优于常用的机器学习分类器的。
而且基于搜索的方法,在某些方面也有自己独特的优势。比如说当我们产生新的参考数据的时候,直接添加到数据库中,就可以完成数据库的升级来进一步提高我们判断的效果。而预先训练的模型,在有新的参考加入的时候,可能需要对模型进行修改,甚至重新训练。尤其是数据量急剧增长的情况下,这个训练过程是非常缓慢的。
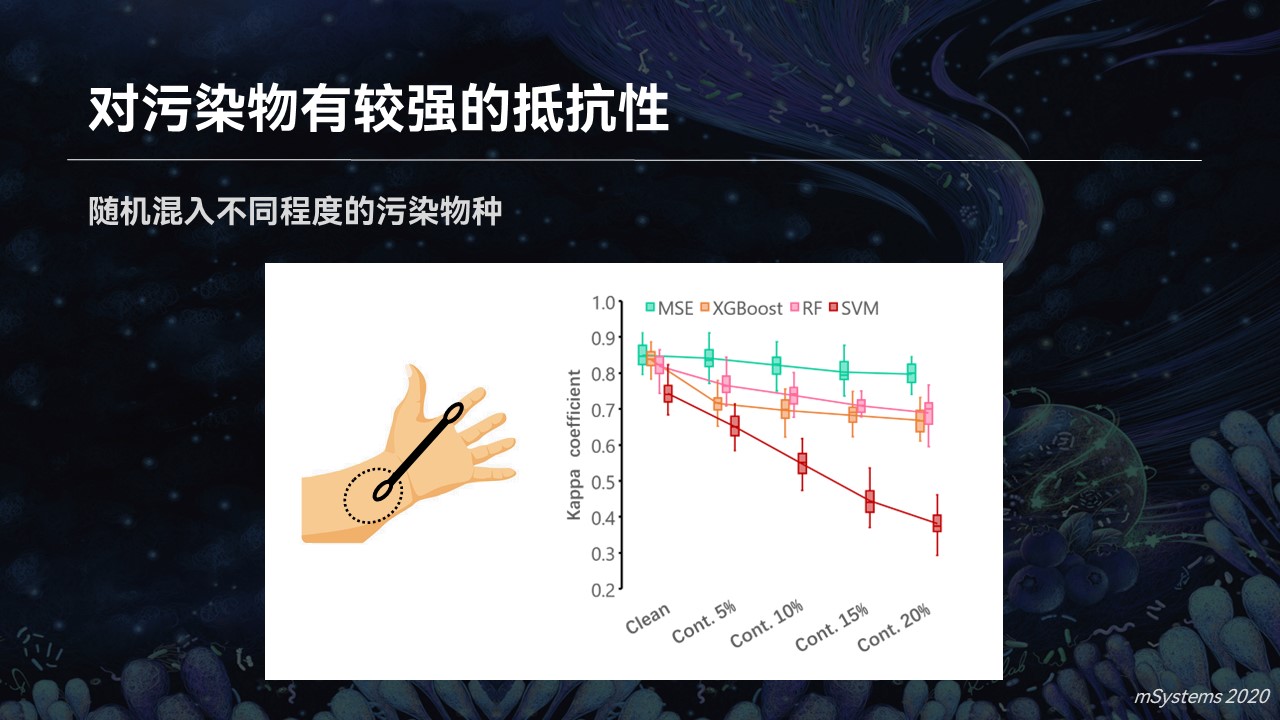
此外,随着菌群检测的进一步普及,我们的采样过程、采样环境不仅限于实验室的环境,有可能是由用户自行完成的。
比如我们在家里用采样的设备进行取样,我们知道生活环境中微生物是几乎无处不在的,那么污染就很难避免。比如我们不小心把采样拭子掉在地上,或者用我们的手指触碰了拭子,会引入污染的物种。
当我们在实验过程中,人为约混入高达20%的污染的时候,我们可以看到,搜索引擎仍然能够很准确地判断出不同的疾病类型。因为它依赖于菌群整体的结构特征和物种之间的亲缘关系,而机器学习的分类器准确度下降的就比较快。
所以利用大数据和搜索引擎,我们提供了菌群检测中一些问题的新的解决方案,包括如何来判断一个菌群是不是健康,以及如果它不健康,最有可能是什么具体的疾病类型。
大数据畅想
在这几年,大家也都知道,人工智能、大数据以及微生物组技术正在发生着翻天覆地的变化。我们都很幸运,是这些发展的见证者、受益者,甚至是推动者。
所以,我作为一个计算机背景的生物信息从业者,也想稍微跟大家分享一下,我对于目前以及以后的微生物组技术的一些机遇和挑战的思考。
首先,我们可能面临更多的数据类型。
目前的菌群研究多数依赖于各种不同的测序类型,通过对测序序列的分析知道了群落中有什么、能干什么。而近期中科院青能所徐健教授团队开发的拉曼组技术,有望能够提供一个实时级的菌群检测,让我们知道群落里面有什么、能干什么、正在做什么。
与此同时,微生物组的技术想要进一步地落地应用,也要解决一些复杂应用场景的问题。
我们在以往的研究中建了很多研究队列,要考虑到尽可能地让背景清晰、避免一些混杂因素的干扰,专注于群落结构功能与一些特定疾病之间的互作。因此在设计队列的时候,往往让每位受试者都有一个明确的标签,比如说是健康对照,或者说一个具体的疾病类型。
然而在实际的检测过程中,情况要复杂的多。特别是在一些中老年队列或者说慢性病患者队列中,一个患者可能深受数种疾病的困扰。这些共存病或并发症共同与肠道菌群相互作用,会严重干扰状态的识别。所以,我们需要开发更先进的技术来克服这些困难。
此外,大数据的模型也有可能为我们提供对微生物组技术新的认识。举个例子,我们常用的网页搜索引擎大多数是依赖于关键字的匹配和判断,而ChatGPT除了字面意思之外,还可以从语义方面去理解用户的需求。
我们的搜索引擎也是如此,将要面临从菌群结构特征的匹配到群落组成意义的理解。之前我们也做过很有意思的尝试,根据群落的整体特征来判断一个菌群的来源,准确性能够达到90%以上。我们把相同的问题给ChatGPT,它虽然没有受过这方面的专业训练,但是它仍然能够得出类似的结论。
所以,大模型的发展有可能让我们重新认识菌群,获得一些不一样的理解。
最后,我由衷地感谢博士期间的导师徐健教授,以及我的合作者UCSD的Rob KNIGHT教授对我的工作的支持。
谢谢大家!